 Preparato
istologico epatico con emocromatosi . Da www.cmsp.com/vlightbox/vlba544/welcome
Preparato
istologico epatico con emocromatosi . Da www.cmsp.com/vlightbox/vlba544/welcome
(Familial emochromatosis)
Introduzione e definizione.
L’emocromatosi è una condizione patologica caratterizzata da una eccessiva deposizione di ferro nelle cellule parenchimali di diversi organi (poiché il ferro non è sempre di provenienza eritrocitaria, si parla anche più generalmente di siderocromatosi).
In altri casi il ferro non si accumula all’interno delle cellule parenchimali, ma di quelle del sistema reticoloendoteliale; la definizione più corretta è di emosiderosi o anche semplicemente siderosi.
Le cause di emocromatosi possono essere diverse, ma distinguono due forme principali:
emocromatosi primitiva o idiopatica;
emocromatosi secondaria o acquisita, quest’ultima sua volta comprendente:
– emocromatosi eritropoietica;
– emocromatosi da cirrosi epatica etilica;
– emocromatosi da eccessivo apporto di ferro.
Preparato
istologico epatico con emocromatosi . Da www.cmsp.com/vlightbox/vlba544/welcome
Basi genetiche dell’emocromatosi
L’emocromatosi idiopatica, descritta da von Recklingausen nel 1889, è una malattia ereditaria autosomica recessiva e quindi clinicamente evidente soltanto nei soggetti omozigoti. È una patologia rara: nelle popolazioni anglosassoni la forma omozigote ha una prevalenza di circa 0,3% e quella eterozigote di circa il 10%.
Un’altra caratteristica di questa malattia è la sua frequente associazione con alcuni antigeni di istocompatibilità, HLA-A3 soprattutto.
Il difetto ereditario che si pensa sia responsabile della emocromatosi idiopatica sarebbe rappresentato da una alterata regolazione dell’assorbimento intestinale del ferro in rapporto al fabbisogno dell’organismo ed alla disponibilità dei depositi: mentre in un soggetto adulto normale l’assorbimento intestinale di questa sostanza è pari a circa 1 mg/die nell’uomo e a 1,5-2,0 mg/die nella donna, in presenza di emocromatosi idiomatica l’assorbimento intestinale del ferro supera mediamente i 4 mg/die.
Per effetto di questo fatto, legato probabilmente a qualche alterazione della membrana delle cellule della mucosa, viene assorbita una quantità di ferro superiore al fabbisogno dell’organismo: questo provoca, a lungo andare (ci vogliono molti anni), la comparsa di emocromatosi.
Il gene che codifica per la transferrina che lega il ferro a livello intestinale regolandone l’assorbimento è stato identificato sul cromosoma 6 e sono state descritte diverse mutazioni associate all’emocromatosi; le più frequenti sono quella in posizione 845 (più comune nella forma omozigote, con scambio cisteina-tirosina nella sequenza aminoacidica della ceruloplasmina in posizione 282) e quella in 187

Hfe is MHC-related protein that is mutated in the iron- overload disease, hereditary hemochromatosis. Hfe binds to transferrin receptor (tfr) and reduces its affinity for iron-loaded transferrin, implicating hfe in iron metabolism. Da www.cmsp.com/vlightbox/vlbbf13/welcome
La malattia è rarissima prima dei 20 anni e quasi il 75% dei casi si manifesta tra i 40 e i 60, poiché il meccanismo di regolazione dell’assorbimento intestinale di questa sostanza riguarda comunque piccole quantità di ferro e quindi occorre molto tempo prima di raggiungere i livelli che si riscontrano in questi casi (in genere oltre 20 g, ma anche 40-50 g, oltre 10 volte i valori normali compresi tra 3 e 4 g).
Fisiopatologia.
La malattia colpisce soggetti in età adulta o senile ed è molto più frequente nell’uomo che nella donna (5-10:1). Per spiegare questo fatto, è importante tenere conto della periodica perdita mestruale (circa 25-30 mg di ferro), che ritarda o impedisce l’accumulo di ferro nel sesso femminile, e della maggiore frequenza dell’etilismo cronico nel sesso maschile. Quando il ferro è presente nell’organismo in quantità eccessiva, tende a depositarsi sotto forma di ferritina e di emosiderina nelle cellule parenchimali ed in quelle del sistema reticolo-endoteliale di diversi organi e tessuti.
Gli organi che vengono più spesso colpiti in questa malattia sono di seguito specificati.
1. Fegato (circa 95% dei casi): il ferro provoca sofferenza cellulare e progressiva insorgenza di cirrosi, la quale ha le caratteristiche cliniche della classica cirrosi portale, con possibile evoluzione in epatocarcinoma nel 30% circa dei casi.
2. Cute (circa 90% dei casi): la deposizione di ferro determina la comparsa di iperpigmentazione con tipico colorito grigio-ardesia (colore bronzino), dovuto sia all’accumulo di ferro sia, soprattutto, all’aumento del contenuto di melanina in questa sede, come si verifica sempre in caso di emocromatosi e la cui interpretazione è tuttora sconosciuta; nel 10-15% dei casi si riscontra anche iperpigmentazione della mucosa orale, molto più raramente del palato duro e della retina.
3. Pancreas (circa 65% dei casi): il ferro può depositarsi sia nel pancreas esocrino sia nelle isole di Langerhans; nel primo caso determina l’insorgenza di insufficienza pancreatica esocrina (pancreatite cronica), nel secondo caso provoca la distruzione delle cellule insulari e, in particolare, delle cellule _, produttrici di insulina, determinando la comparsa di diabete mellito insulinodipendente.
La triade, cirrosi epatica, iperpigmentazione cutanea e diabete mellito rappresenta il quadro clinico più caratteristico della emocromatosi, la quale, perciò, viene anche denominata diabete bronzino.
Il ferro può depositarsi anche a livello del miocardio (circa 10% dei casi), con compromissione delle cellule miocardiche e comparsa di miocardiopatia dilatativa e scompenso cardiaco oppure di disturbi del ritmo; è inoltre piuttosto comune l’interessamento delle gonadi con sviluppo di ipogonadismo e, quindi, per la particolare prevalenza della emocromatosi nel sesso maschile, di microorchia.
Infine è importante ricordare che nel 25-50% di questi pazienti si riscontra, per ragioni sconosciute, la deposizione non di ferro, ma di cristalli di pirofosfato di calcio (Ca2P2O2 · 2H2O) a livello della sinovia (con comparsa di pseudogotta), oppure delle cartilagini articolari (con insorgenza di condrocalcinosi) o anche del connettivo periarticolare.
Diagnosi.
Nella emocromatosi la sideremia è significativamente aumentata, mentre la capacità totale ferro-legante del plasma (TIBC) è variamente diminuita; inoltre si riscontra sempre, come dato di laboratorio molto importante dal punto di vista diagnostico, l’incremento della ferritina, espressione dell’eccessivo accumulo di ferro nei depositi dell’organismo.
La diagnosi viene fatta, di regola, sulla base delle caratteristiche cliniche di questa patologia (cirrosi epatica, iperpigmentazione cutanea, diabete mellito, cardiomiopatia, artrite, ipogonadismo) e viene confermata mediante il test alla desferrioxamina, che è una sostanza chelante in grado di legare il ferro, favorendone in tal modo l’escrezione urinaria.
La quantità di ferro presente nelle urine risulta proporzionale alle scorte di questa sostanza nell’organismo ed in condizioni normali la sideruria è in genere inferiore, e comunque mai superiore, a 1 mg/24 h.
Decorso e terapia
La prognosi in linea generale è piuttosto severa; le principali cause di morte nei pazienti non trattati sono rappresentate dallo scompenso cardiaco (30%), dall’epatocarcinoma (30%), dalla insufficienza epatica e dalla ipertensione portale (25%).
La terapia è fondata sul periodico ricorso a salassi di 300-500 ml di sangue ciascuno, da ripetersi periodicamente nel tempo per alcuni anni fino alla rimozione dell’eccesso di ferro presente nell’organismo.
Sull’argomento vedi anche:
http://www.emocromatosi.it/
http://www.uildm.org/dm/129/ricerca/42emo.htm
http://www.fleming-research.it/frame-news02.htm
http://www.niddk.nih.gov/health/digest/pubs/hemochrom/hemochromatosis.htm
L’IPERCOLESTEROLEMIA EREDITARIA
(Familial Hypercholesterolaemia, FH, IF)
Introduzione e definizione.
L’Ipercolesterolemia Ereditaria o Familiare è una malattia genetica a carattere autosomico dominante che si osserva nella forma eterozigote in circa 1 su 500 casi, mentre nella forma omozigote, più rara, la prevalenza è di circa 1 su 1.000.000. Nei pazienti con IF i livelli di colesterolo totale nel plasma e di colesterolo LDL sono elevati fin dalla nascita e rimangono tali per tutta la vita. Negli adulti non trattati i livelli di colesterolo totale variano da 325 a 450 mg/dL per i soggetti eterozigoti, e da 500 a 1000 mg/dL negli omozigoti, contro i normali valori plasmatici di colesterolo che si aggirano intorno ai 170 – 200 mg/dL , pur mantenendo normali i valori plasmatici di trigliceridi e normali o ridotti i valori di colesterolo HDL.[ 1]
Basi genetiche dell’Ipercolesterolemia Familiare.
Studi sul DNA hanno rivelato che a causare l’Ipercolesterolemia Familiare è la mutazione del gene che codifica il recettore transmembrana per le lipoproteine LDL nelle cellule epatiche o degli altri tessuti interessati alla clearence delle LDL plasmatiche.
Il gene responsabile si trova nel cromosoma 19 ed è composto da 45.000 paia di basi con 18 esoni. Essi codificano per una glicoproteina di membrana di 772 aminoacidi che consta di 5 domini funzionali.
A tutt’oggi sono state descritte circa 350 mutazioni responsabili dell’alterata produzione del recettore per le LDL, suddivise per classi a seconda del dominio funzionale che compromettono (fig.1) [ 2]
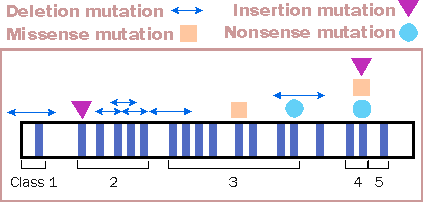
Fig.1Examples of mutations in the cholesterol-related LDL receptor gene. Shown are the 18 exons of the gene and various combinations of known mutations, classes 1-5. (This illustration is adapted from a more detailed diagram in J.L. Goldstein and M.S. Brown, "Familial Hypercholesterolemia," in C.R. Scriver , A.L. Beaudet, W.S. Sly, and D. Valle, editors, The Metabolic Basis of Inherited Disease, 6th edition (New York: McGraw-Hill, 1989), p. 1232.) [ 2]
Nella prima classe di mutazioni il recettore non viene sintetizzato.Nella seconda classe il recettore è sintetizzato, ma non viene espresso sulla membrana cellulare.Nella terza classe di mutazioni il recettore è presente ma non riesce a legare le LDL; nella quarta classe non riesce a stare in membrana, mentre nella quinta classe di mutazioni esso è presente in membrana, si lega alle LDL, ma non le riesce ad endocitare all’interno del citoplasma.
Fisiopatologia.
Brown e Goldstein hanno dimostrato che i pazienti con IF presentano o assenza del recettore per le LDL nelle cellule epatiche, o presenza di recettori per le LDL ma difettosi e con attività ridotta a meno del 10% del normale, per ultimo presenza di recettori normali ma incapaci di mediare l’endocitosi delle LDL.
Normalmente, ogni giorno, circa il 45% delle LDL plasmatiche è rimosso sia per opera del fegato, sia dai tessuti extra epatici, specialmente ghiandole surrenali e tessuto adiposo (circa un 50% dai tessuti extra epatici, il rimanente dal fegato).
|
I recettori per le LDL (LDL – r) (fig 2) sono glicoproteine di membrana; ogni cellula può contenerne da 15.000 a 70.000. Essi captano il colesterolo LDL proveniente dal circolo ematico riconoscendo e legando la componente proteica Apo –B 100 delle LDL (recettori specifici Apo – B 100). Una volta avvenuto il legame le LDL sono inglobate mediante endocitosi e indirizzate verso i lisosomi con cui vengono fuse. Le particelle di LDL vengono quindi degradate da proteasi e lipasi acide lisosomiali. Il colesterolo diffonde dal lisosoma e blocca così la propria sintesi epatica (blocco con meccanismo di feed-back negativo dell’enzima βidrossi-βmetil-glutarilCoA-reduttasi) e va ad attivare l’enzima aciltransferasi che forma esteri del colesterolo. (fig.3)
L’attività del recettore per le LDL è determinata da fattori nutrizionali, ormonali e genetici, dovuti al bisogno intracellulare di colesterolo. Quando il colesterolo è captato intracellularmente blocca, con un meccanismo di feed-back negativo, la produzione dei recettori delle LDL inibendo quindi la propria eccessiva assunzione, portando all’espressione di una proteina SRE (sterol response element) che agisce sulla regione "promoter" del gene per gli LDL-r, disattivandolo. Per contro l’attività dei recettori per le LDL è stimolata dall’insulina e dagli ormoni tiroidei. [ 3]
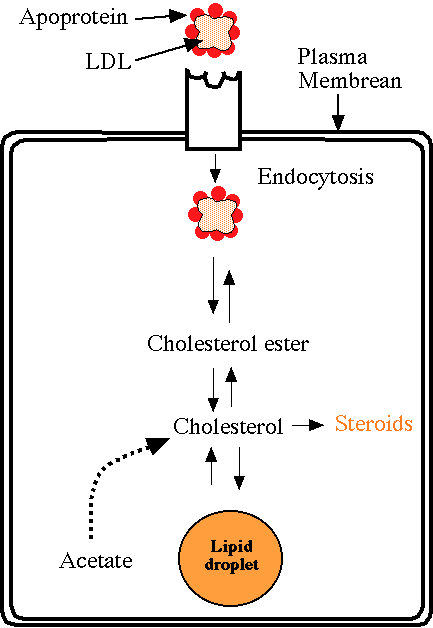
Fig.3 Trasporto e metabolismo del colesterolo
Sintomatologia clinica.
La ridotta attività degli LDL-r porta, fisiologicamente, all’aumento di altri meccanismi interessati all’eliminazione del colesterolo LDL, normalmente coinvolti per il 15% della totale rimozione delle LDL dal circolo sanguigno. Tali meccanismi interessano specifici recettori per le LDL modificate chimicamente (per es. ossidate) presenti sui macrofagi (scavenger receptors) del sangue. Una volta captate dai macrofagi le LDL ossidate provocano la morte dei loro stessi assalitori, portando, con una serie di processi dovuti ad un danno endoteliale, che vanno dalla necrosi all’infiammazione, alla formazione della placca ateromasica che causa a sua volta disturbi cardiovascolari. [ 3]
I pazienti con IF sviluppano coronaropatie aggressive e precoci (fin dall’infanzia nei casi omozigoti, in età giovanile –30/40anni – negli eterozigoti); xantomi in età adulta per gli eterozigoti e fin dall’infanzia per gli omozigoti (deposito di colesterolo in diverse sedi: tendine di Achille, palpebre, gomiti). (fig.4)
Manifestazioni secondarie da IF sono ipotiroidismo, malattia ostruttiva a carico del fegato e sindrome nefrotica.
I danni più seri sono quelli a carico del cuore e del sistema vascolare, che possono portare a morte improvvisa; tuttavia la diagnosi di Ipercolesterolemia Familiare non pone particolari problemi in considerazione della giovane età dei pazienti, degli eccessivi livelli della colesterolemia e della positività della storia familiare.
La prevenzione dei danni dell’IF si basa sull’anamnesi della storia familiare, sull’informazione genetica e sull’allontanamento dei fattori di rischio quali ipertensione, fumo di sigaretta, ecc., nonché sull’importanza fondamentale della dieta ipolipidica; ma nulla di tutto ciò sarebbe utile al paziente se non opportunamente integrato con uno screening periodico dei valori plasmatici di colesterolo totale e di colesterolo LDL, da una terapia farmacologia mirata alla rimozione delle lipoproteine LDL dal plasma. A volte l’unica soluzione resta il trapianto di fegato.
Sull’argomento vedi anche:
http://www.aging.cnr.it/uo/uo3_114.htm
http://www.cardiolink.it/PR-BG-colesterolo.htm
